

Ijraset Journal For Research in Applied Science and Engineering Technology
Homology Modeling and Structure Based Drug Design for Human Gastric Cancer
Authors: Uma Kumari, Manaswinee Bora, MN. Akshaya
DOI Link: https://doi.org/10.22214/ijraset.2023.56165
Certificate: View Certificate
Abstract
This research article delves into the multifaceted world of structural biology and bioinformatics, exploring a diverse array of tools and resources employed to decipher the intricacies of proteins and their implications in health and disease. The paper navigates through various computational tools and databases used for sequence analysis, structural analysis, mutation site detection, and molecular docking, shedding light on their individual contributions in unraveling the secrets of proteins. COBALT serves as the initial compass, guiding researchers in the alignment of protein sequences, identifying conserved domains, and elucidating structural features in proteins. The exploration continues, with a focus on tools such as RasMol, PyMol, and Pharmit. This research article explores a range of bioinformatics tools and databases crucial for elucidating the complex world of proteins and their involvement in diseases like gastric cancer. It begins by highlighting the pivotal role of the Catalogue of Somatic Mutations in Cancer (COSMIC) in mutation site detection, providing a comprehensive repository of somatic mutations in cancer. The Kyoto Encyclopedia of Genes and Genomes (KEGG) complements this with a wealth of biological data and pathways. Shifting the focus to protein stability and dynamics, the article introduces DynaMUT, a computational tool employing molecular dynamics simulations to predict mutation impacts on protein behavior. Lastly, SwissDock takes the stage for drug discovery, facilitating the prediction and analysis of interactions between small molecules and target proteins. Collectively, these tools and databases offer an interdisciplinary lens through which researchers unravel protein intricacies and their relevance in diseases, exemplifying the indispensable role of structural biology and bioinformatics in advancing biological understanding.
Introduction
I. INTRODUCTION
The structure of recombinant human gastric lipase (rHGL) includes a core domain and a "cap" domain. It possesses a classical catalytic triad and N-glycosylation sites. HGL plays a crucial role in newborns, as pancreatic lipase is not fully developed in infants, and it initiates the digestion of dietary fats. Gastric lipase and other preduodenal lipases are resistant to acidic conditions and can assist in fat digestion in the stomach and duodenum. Human lysosomal acid lipase (HLAL) is similar in sequence to HGL but has different enzymatic activities, including the hydrolysis of triglycerides and cholesteryl esters. Mutations in the LIPA gene(Lysosomal Acid Lipase A gene),which encodes HLAL, can lead to genetic disorders like Wolman disease and cholesteryl ester storage disease. Recombinant HGL is produced using the baculovirus/insect cell system and has high specific activity. Crystals of rHGL are obtained and analyzed for their structure, revealing specific characteristics. The catalytic activity of rHGL is tested both in dissolved and intact crystals, with specific activity measured. In situ catalytic activity is assessed by incubating a single rHGL crystal in a substrate solution, resulting in coloration [1].HGL is an enzyme secreted by the stomach that remains stable and active in the highly acidic environment of the stomach. It plays a significant role in the digestion of dietary fats, contributing to approximately 30% of the fat digestion processes in humans. HGL is described as a globular protein belonging to the α/β hydrolase fold family. Its catalytic serine is located deep within the enzyme and is covered by a domain known as the "extrusion domain." This extrusion domain comprises a "cap" domain and a segment consisting of 58 residues referred to as the "lid”. The exact roles played by the cap and lid domains during the catalytic process of HGL have not been fully understood. To investigate these roles, the crystal structure of the open form of dog gastric lipase (DGL) in complex with a covalent inhibitor was solved. The inhibitor and detergent molecules were used to mimic a triglyceride substrate, interacting with residues from both the cap and lid domains [2].
Gastric cancer, also known as stomach cancer, remains a significant global health concern, accounting for a substantial portion of cancer-related morbidity and mortality. It poses a particularly formidable challenge due to its often asymptomatic early stages and the limited effectiveness of current therapeutic options. As such, the exploration of novel treatment strategies, including structure-based drug design, has garnered increasing attention in the quest to combat this disease.
One of the critical aspects of developing targeted therapies for gastric cancer involves understanding the molecular structures and interactions of proteins associated with the disease. In this context, homology modeling and structure-based drug design play pivotal roles. Homology modeling, a computational technique, enables the prediction of protein structures based on the known structures of homologous proteins. This method has proven invaluable in elucidating the three-dimensional architecture of biologically significant proteins, including those implicated in gastric cancer. Human gastric lipase (1HLG) is one such protein that has emerged as a potential target for the development of novel therapeutic interventions in gastric cancer. Gastric lipase, primarily associated with lipid digestion in the stomach, has been found to have diverse biological roles, including implications in tumor progression and cancer metabolism. Understanding the structural characteristics of human gastric lipase (1HLG) and its potential druggable sites is essential for the rational design of targeted therapies aimed at mitigating the effects of gastric cancer [3,4,5].
Gastric cancer is a global health concern characterized by its high prevalence, particularly in certain regions such as East Asia, South America, and parts of Eastern Europe. It ranks as the fifth most common cancer worldwide and the third leading cause of cancer-related mortality, making it a significant public health issue [6]. Gastric cancer exhibits marked geographic and demographic variations in its incidence rates. Understanding these variations and the associated risk factors, including Helicobacter pylori infection, dietary habits, and genetic predisposition, is crucial in developing effective prevention and treatment strategies. The Tripartite Motif (TRIM) family of proteins, encoded by TRIM genes, is known for their role in various human diseases, including cancer. TRIM16 is one such protein, with tumor suppressive properties. TRIM16 is associated with the retinoid signaling pathway, which plays a role in cancer cell death. Overexpression of TRIM16 has been linked to decreased cell growth, motility, and tumorigenicity in various cancers [7]. Helicobacter pylori infection is a well-established risk factor for gastric cancer. This bacterium colonizes the stomach lining and can lead to chronic gastritis, peptic ulcers, and eventually, in some cases, gastric cancer. Understanding the molecular mechanisms by which H. pylori promotes carcinogenesis, such as through the activation of inflammatory pathways and the disruption of gastric epithelial homeostasis, is crucial for developing preventive strategies and targeted therapies [8].The asymptomatic nature of early-stage gastric cancer and the lack of highly sensitive screening tools pose significant diagnostic challenges. Consequently, gastric cancer often goes undetected until it reaches an advanced stage, limiting treatment options and reducing overall survival rates. Proteins are key players in cellular functions, and alterations in their structure and function are often associated with cancer initiation and progression. Mutations, overexpression, or dysregulation of specific proteins can contribute to carcinogenesis. Determining the three-dimensional structures of proteins involved in gastric cancer provides crucial insights into their mechanisms of action. Structural data can reveal active sites, binding pockets, and potential druggable regions, aiding in the rational design of targeted therapies. Homology modeling relies on the premise that evolutionarily related proteins share structural similarities. This technique involves sequence alignment, template selection, and model building, ultimately resulting in a predicted protein structure. The advent of precision medicine allows for the tailoring of treatments to the genetic and molecular profiles of individual patients. Targeted therapies designed using protein structures offer the potential for more effective and less toxic interventions. While personalized medicine holds great promise, there are challenges to overcome, including the identification of appropriate biomarkers and the development of novel therapeutics [9,10,11,12].
II. MATERIAL AND METHODS
For the sequence analysis of protein COBALT has been used. COBALT is a bioinformatics tool developed by the National Center for Biotechnology Information (NCBI) for the alignment of protein sequences. It is primarily used for identifying and comparing conserved domains and structural features in proteins. Cobalt is a part of the broader suite of tools and resources offered by NCBI to help researchers analyze and interpret biological data. Cobalt is designed to align protein sequences. It can be used to identify regions of similarity or conservation among a set of protein sequences, which is critical for understanding the relationships between different proteins and their functional domains [13,14].
For structural analysis of protein RasMol, PyMol, Pharmit has been used. RasMol is an early molecular visualization software tool that has been widely used in the field of structural biology and biochemistry. It was first developed in the early 1990s by Roger Sayle and has been instrumental in the exploration of protein and nucleic acid structures [15,16]. PyMOL is a more modern and versatile molecular visualization tool that has gained popularity in the field of structural biology. It was created by Warren L. DeLano and is known for its user-friendly graphical user interface and scripting capabilities. PyMOL provides high-quality molecular rendering with advanced graphics capabilities, allowing users to create visually appealing images and animations of molecular structures [17,18].Pharmit is a molecular docking and virtual screening tool used for the computational analysis of protein-ligand interactions.
It is designed to help researchers explore the binding of small molecules (ligands) to protein targets in the context of drug discovery and molecular modeling. Pharmit is particularly useful for identifying potential drug candidates and understanding the binding affinity of ligands to target protein [19].
For detecting mutation sites in the protein COSMIC, DynaMut and KEGG has been used. COSMIC (Catalogue of Somatic Mutations in Cancer) is a comprehensive and widely recognized database that compiles and curates information on somatic mutations in cancer. It is an essential resource for researchers, oncologists, and clinicians who seek to understand the genetic alterations that drive cancer development and progression. COSMIC primarily focuses on somatic mutations, which are genetic alterations that occur in a person's cells during their lifetime and contribute to the development of cancer. These mutations can be in the form of single nucleotide variants (SNVs), insertions, deletions, copy number variations (CNVs), and structural variants [20,21].KEGG, or the Kyoto Encyclopedia of Genes and Genomes, is a comprehensive bioinformatics resource that provides valuable information on a wide range of biological data, with a primary focus on genomics, pathways, diseases, and drugs. KEGG is a well-established and widely used resource in the fields of molecular biology, genetics, bioinformatics, and systems biology. It offers various tools, databases, and knowledge bases to help researchers explore and analyze biological processes [22,23]. DynaMUT is a computational tool and web server designed for the analysis and prediction of the impacts of mutations on protein stability, dynamics, and function. This tool is particularly useful in the field of structural biology, bioinformatics, and protein engineering. DynaMUT employs molecular dynamics simulations and various computational techniques to evaluate how mutations in a protein sequence can affect its stability and functional properties [24].
For docking purpose SwissDock has been utilized. SwissDock is a powerful and widely used molecular docking software and web server that is designed to predict and analyze the interactions between small molecules (ligands) and target proteins. Molecular docking is a computational technique used in drug discovery, bioinformatics, and structural biology. SwissDock has been developed by the Molecular Modeling Group at the University of Lausanne in Switzerland [25].





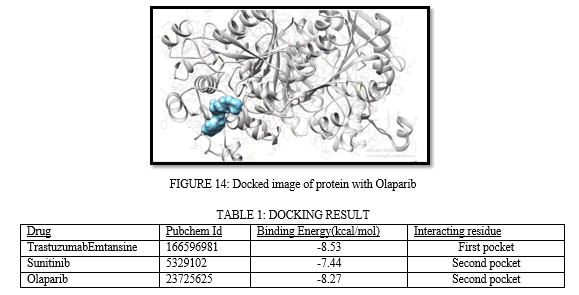
TrastuzumabEmtansine, Sunitinib, and Olaparib are three important drugs that have significant roles in various aspects of cancer treatment and targeted therapy. TrastuzumabEmtansine is an antibody-drug conjugate used in the treatment of HER2-positive breast cancer. Sunitinib is a tyrosine kinase inhibitor employed in the management of advanced kidney cancer, gastrointestinal stromal tumors (GISTs), and other malignancies. Olaparib is a PARP inhibitor used in the treatment of ovarian and breast cancers, particularly those associated with BRCA mutations.TrastuzumabEmtansine, often referred to as T-DM1, is an antibody-drug conjugate. It combines the monoclonal antibody trastuzumab with the cytotoxic agent DM1 (emtansine). Trastuzumab targets the HER2 receptor, which is overexpressed in some breast cancers, while DM1 is a potent chemotherapy agent. Sunitinib is a tyrosine kinase inhibitor (TKI) that interferes with the signaling pathways involved in cell proliferation and angiogenesis. It targets multiple receptor tyrosine kinases, including those involved in vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) signalling. Olaparib is a poly (ADP-ribose) polymerase (PARP) inhibitor. It interferes with the repair of DNA in cancer cells, particularly those with BRCA mutations. By inhibiting PARP, Olaparib prevents the repair of DNA damage, leading to the death of cancer cells.
The utilization of these drugs could be explored in the framework of structure-based drug design and personalized medicine for gastric cancer. Their interactions with specific protein targets or mutation profiles may play a critical role in developing novel therapeutic interventions.
Conclusion
In conclusion, this research article offers a comprehensive exploration of the tools, databases, and drugs that collectively form the foundation for understanding the intricate world of proteins and their implications in the context of gastric cancer. The use of essential bioinformatics tools like COSMIC and KEGG empowers researchers to identify somatic mutations and comprehend the genetic underpinnings of cancer progression. Furthermore, the application of computational tools such as DynaMUT allows for the prediction of how mutations can impact protein behavior, a critical aspect of structural biology and protein engineering. To complete this holistic journey, the article spotlights SwissDock, a potent platform for drug discovery, enabling the prediction and analysis of interactions between small molecules and target proteins. Notably, the inclusion of TrastuzumabEmtansine, Sunitinib, and Olaparib underscores their significance in the realm of cancer treatment, especially within the context of personalized medicine and structure-based drug design for gastric cancer. These drugs, each with their unique mechanisms and targets, hold promise in advancing the field of oncology and offering tailored solutions for patients. The interdisciplinary nature of structural biology and bioinformatics, as showcased in this article, emerges as a cornerstone in advancing our understanding of complex biological processes and shaping the future of precision medicine in the fight against cancer.
References
[1] Alain Roussel, Stéphane Canaan, Marie-Pierre Egloff, MireilleRivière, Liliane Dupuis, Robert Verger, Christian Cambillau, Crystal Structure of Human Gastric Lipase and Model of Lysosomal Acid Lipase, Two Lipolytic Enzymes of Medical Interest*, Journal of Biological Chemistry, Volume 274, Issue 24, 1999, Pages 16995-17002, ISSN 0021-9258, https://doi.org/10.1074/jbc.274.24.16995. [2] Miled, N., Bussetta, C., De caro, A., Rivière, M., Berti, L., & Canaan, S. (2003). Importance of the lid and cap domains for the catalytic activity of gastric lipases. Comparative biochemistry and physiology. Part B, Biochemistry & molecular biology, 136(1), 131–138. https://doi.org/10.1016/s1096-4959(03)00183-0 [3] Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C., &Lordick, F. (2020). Gastric cancer. The Lancet, 396(10251), 635-648. [4] Van Cutsem, E., Sagaert, X., Topal, B., Haustermans, K., &Prenen, H. (2016). Gastric cancer. The Lancet, 388(10060), 2654-2664. [5] Correa, P. (2013). Gastric cancer: overview. Gastroenterology Clinics, 42(2), 211-217. [6] Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., &Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 68(6), 394–424. https://doi.org/10.3322/caac.21492 [7] Afshar, J., Mehrzad, J., Mehrad-Majd, H., Goshayeshi, L., &Saeidi, J. (2021). Prognostic Significance of Tripartite Motif Containing 16 Expression in Patients with Gastric Cancer. Asian Pacific journal of cancer prevention : APJCP, 22(8), 2445–2451. https://doi.org/10.31557/APJCP.2021.22.8.2445 [8] Loor, A., &Dumitra?cu, D. L. (2016). Helicobacter pylori infection, gastric cancer and gastropanel. Rom J Intern Med, 54(3), 151-156. [9] Zhang, J., Lu, C., Shang, Z., Xing, R., Shi, L., &Lv, Y. (2012). p42. 3 gene expression in gastric cancer cell and its protein regulatory network analysis. Theoretical biology and medical modelling, 9(1), 1-10. [10] Futami, T., Kawase, T., Mori, K., Asaumi, M., Kihara, R., Shindoh, N., & Kuromitsu, S. (2019). Identification of a novel oncogenic mutation of FGFR4 in gastric cancer. Scientific Reports, 9(1), 14627. [11] Aktas, S. H., Taskin-Tok, T., Al-Khafaji, K., & Ak?n-Bal?, D. F. (2022). A detailed understanding of the COL10A1 and SOX9 genes interaction based on potentially damaging mutations in gastric cancer using computational techniques. Journal of Biomolecular Structure and Dynamics, 40(22), 11533-11544. [12] Wuputra, K., Ku, C. C., Kato, K., Wu, D. C., Saito, S., & Yokoyama, K. K. (2021). Translational models of 3-D organoids and cancer stem cells in gastric cancer research. Stem Cell Research & Therapy, 12(1), 1-16. [13] Tripathi, Anurag&Kumari, Uma. (2023). Comparative Analysis and Drug Target Identification of Human Kidney Cancer. International Journal for Research in Applied Science and Engineering Technology. 11. 10.22214/ijraset.2023.55777. [14] Kumari, Uma &Tanwar, Aastha& George, Jositta&Nayak, Daityari. (2023). NGS Analysis to Detect Mutation in Brain Tumor Diagnostic. International Journal for Research in Applied Science and Engineering Technology. 11. 10.22214/ijraset.2023.54895. [15] Kumari, Uma &Tripathi, Kartik. (2023). Computational Analysis and Molecular Docking Approach for Liver Cancer. International Journal for Research in Applied Science and Engineering Technology. 11. 1828 to 1633. 10.22214/ijraset.2023.54927. [16] Pikora, M., &Gieldon, A. (2015). RASMOL AB - new functionalities in the program for structure analysis. ActabiochimicaPolonica, 62(3), 629–631. https://doi.org/10.18388/abp.2015_972 [17] Seeliger, D., & de Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/Vina. Journal of computer-aided molecular design, 24(5), 417–422. https://doi.org/10.1007/s10822-010-9352-6 [18] Mura, C., McCrimmon, C. M., Vertrees, J., &Sawaya, M. R. (2010). An introduction to biomolecular graphics. PLoS computational biology, 6(8), e1000918. https://doi.org/10.1371/journal.pcbi.1000918 [19] Koes D. R. (2018). The Pharmit Backend: A Computer Systems Approach to Enabling Interactive Online Drug Discovery. IBM journal of research and development, 62(6), 1–6. https://doi.org/10.1147/jrd.2018.2883977 [20] Forbes, S. A., Beare, D., Gunasekaran, P., Leung, K., Bindal, N., Boutselakis, H., Ding, M., Bamford, S., Cole, C., Ward, S., Kok, C. Y., Jia, M., De, T., Teague, J. W., Stratton, M. R., McDermott, U., & Campbell, P. J. (2015). COSMIC: exploring the world\'s knowledge of somatic mutations in human cancer. Nucleic acids research, 43(Database issue), D805–D811. https://doi.org/10.1093/nar/gku1075 [21] Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., Boutselakis, H., Cole, C. G., Creatore, C., Dawson, E., Fish, P., Harsha, B., Hathaway, C., Jupe, S. C., Kok, C. Y., Noble, K., Ponting, L., Ramshaw, C. C., Rye, C. E., Speedy, H. E., … Forbes, S. A. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic acids research, 47(D1), D941–D947. https://doi.org/10.1093/nar/gky1015 [22] Kanehisa, M., &Goto, S. (2000). KEGG: kyotoencyclopedia of genes and genomes. Nucleic acids research, 28(1), 27–30. https://doi.org/10.1093/nar/28.1.27 [23] Minoru Kanehisa, Miho Furumichi, Mao Tanabe, Yoko Sato, KanaeMorishima, KEGG: new perspectives on genomes, pathways, diseases and drugs, Nucleic Acids Research, Volume 45, Issue D1, January 2017, Pages D353–D361, https://doi.org/10.1093/nar/gkw1092 [24] Rodrigues, C. H., Pires, D. E., &Ascher, D. B. (2018). DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic acids research, 46(W1), W350–W355. https://doi.org/10.1093/nar/gky300 [25] Grosdidier, A., Zoete, V., &Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic acids research, 39(Web Server issue), W270–W277. https://doi.org/10.1093/nar/gkr366
Copyright
Copyright © 2023 Uma Kumari, Manaswinee Bora, MN. Akshaya. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET56165
Publish Date : 2023-10-15
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online